Tutorial¶
To further demonstrate the functionality of Parsnp we have prepared two small tutorial datasets. The first dataset is a MERS coronavirus outbreak dataset involving 49 isolates. The second dataset is a selected set of 31 Streptococcus pneumoniae genomes. Both of these datasets should run on modestly equipped laptops in a few minutes.
49 MERS Coronavirus genomes
Download genomes:
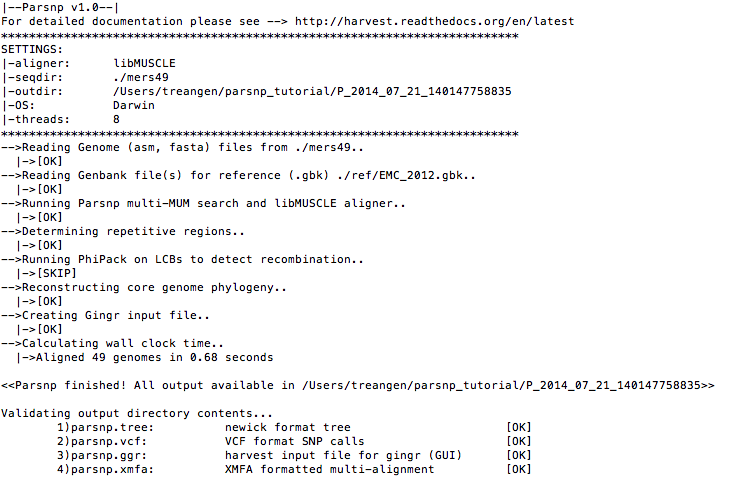
Run parsnp with default parameters
parsnp -g ./mers_virus/ref/England1.gbk -d ./mers_virus/genomes -cCommand-line output
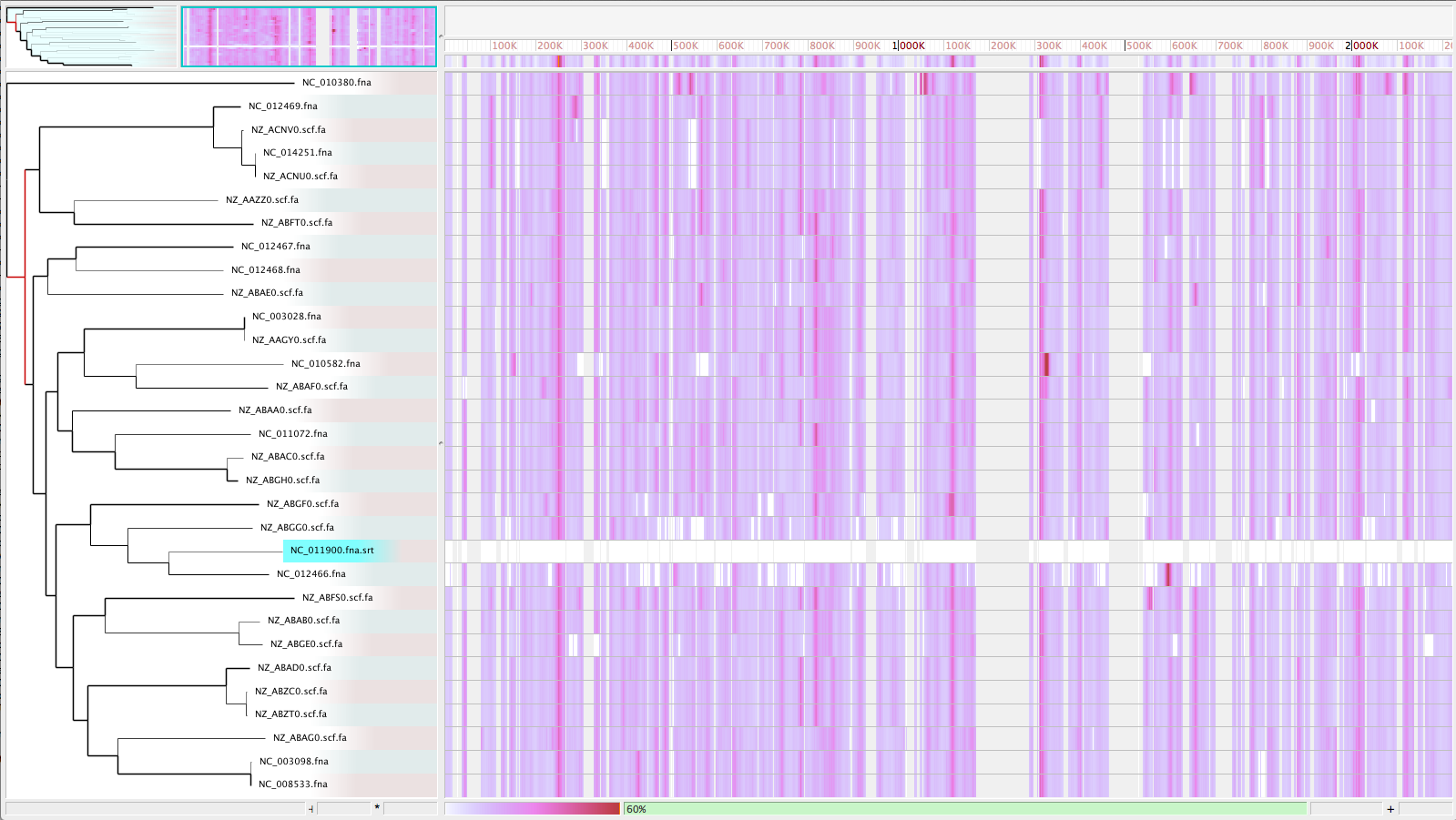
- Visualize with Gingr
GGR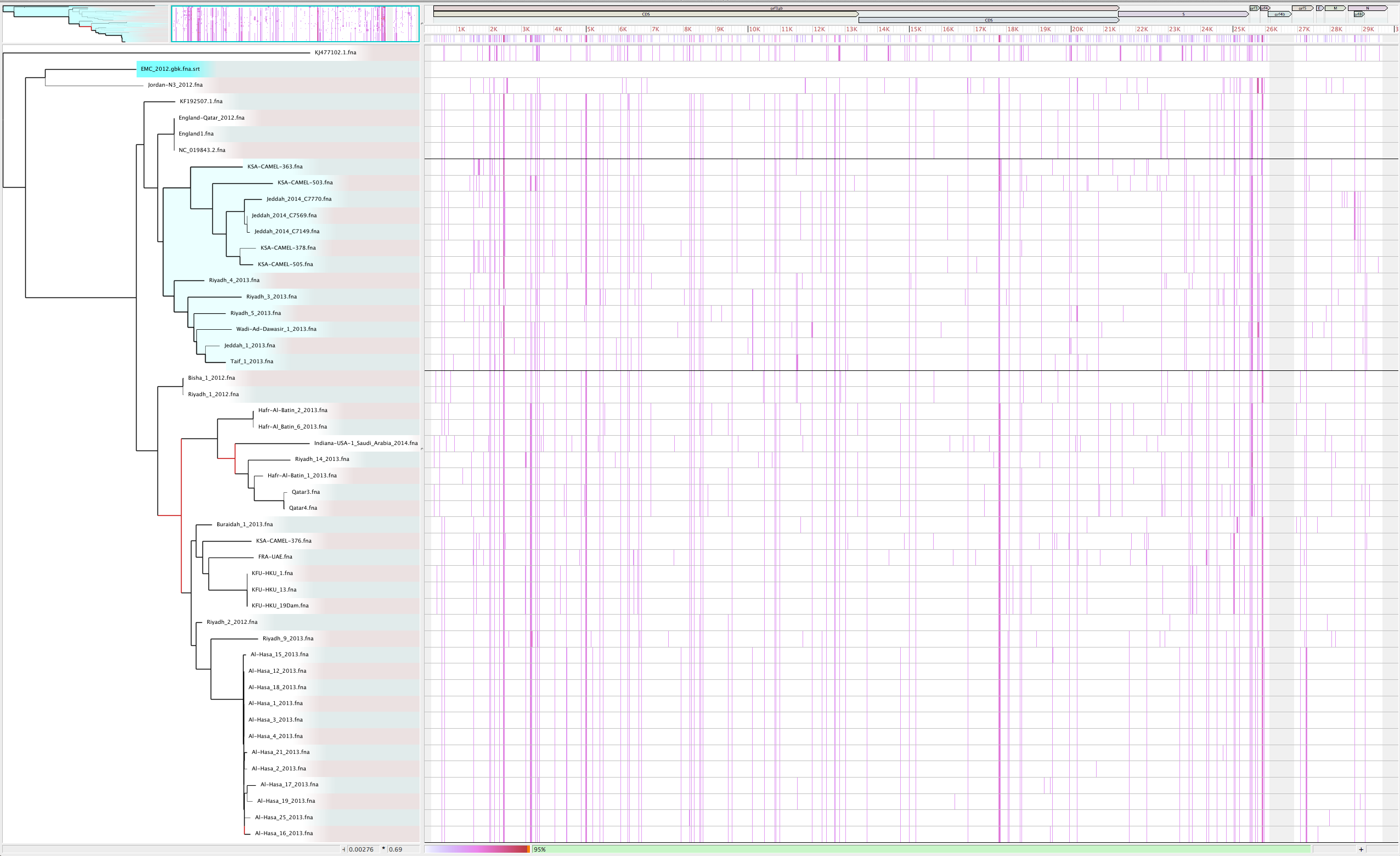
Configure parameters
95% of the reference is covered by the alignment. This is <100% mainly due to a 1kbp unaligned region from 26kbp to 27kbp.
To force alignment across large collinear regions, use the -C maximum distance between two collinear MUMs:
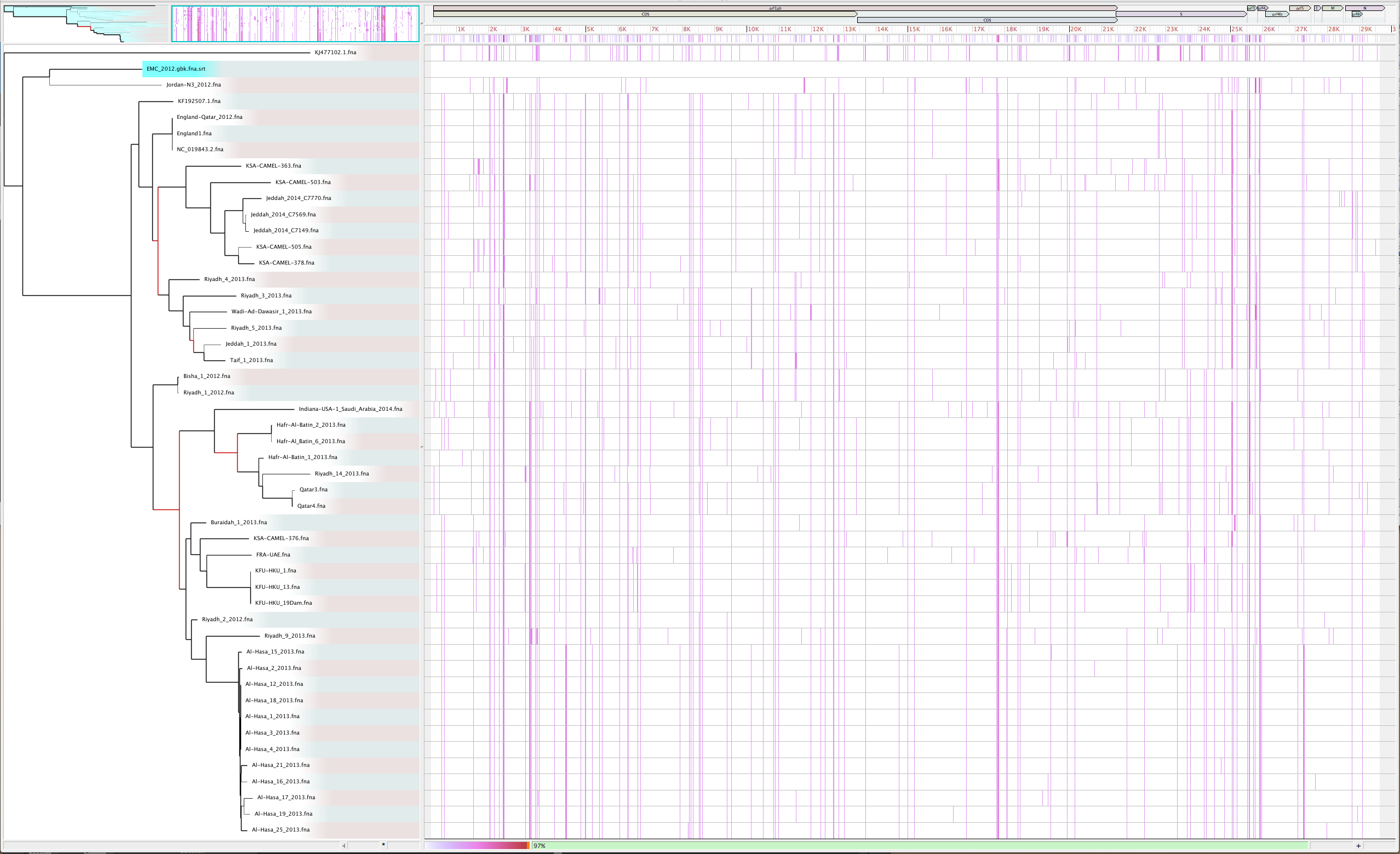
parsnp -g ./mers_virus/ref/England1.gbk -d ./mers_virus/genomes -C 1000 -cVisualize again with Gingr
GGR
- By adjusting the -C parameter, this region is no longer unaligned, boosting the reference coverage to 97%.
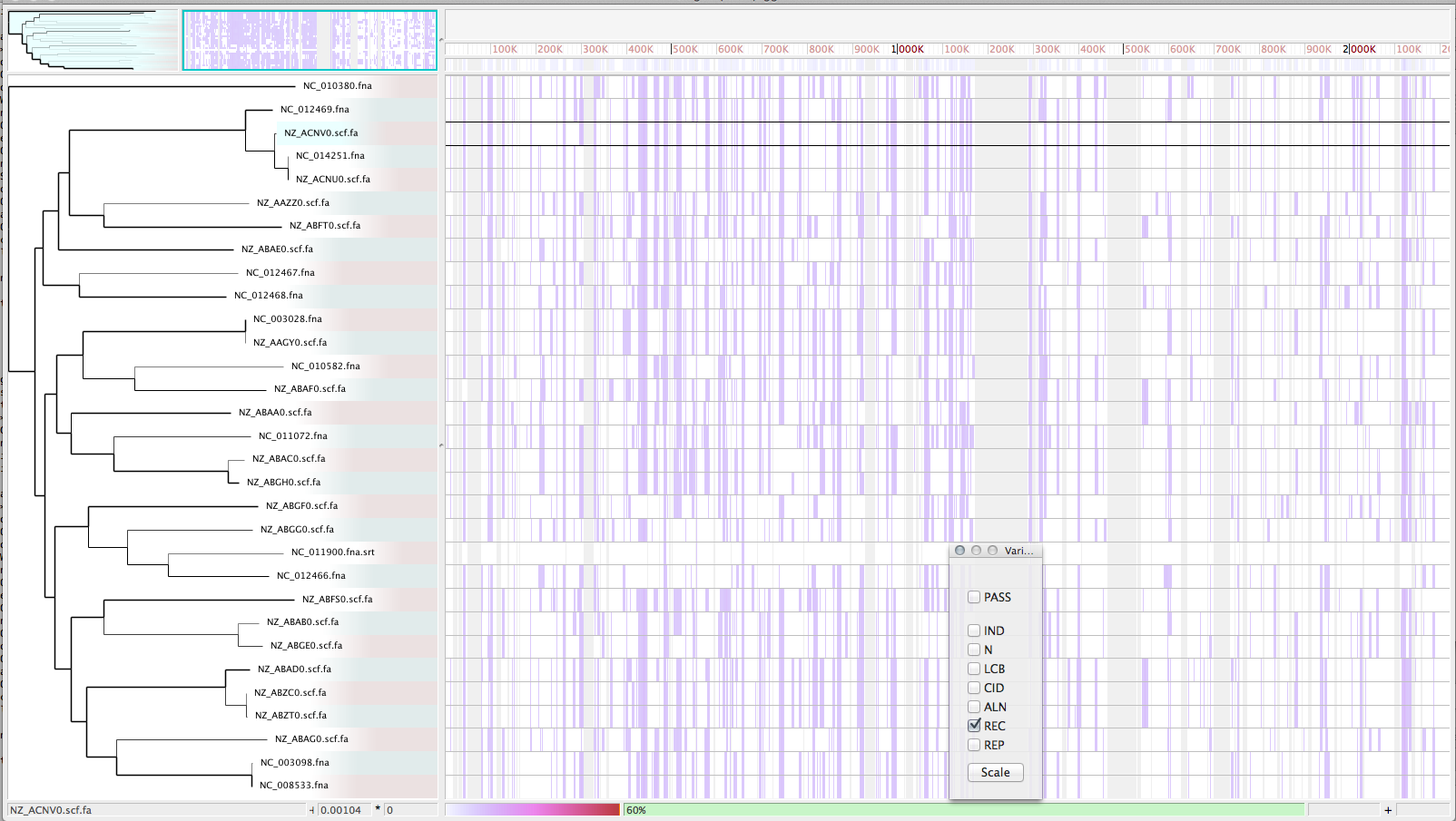
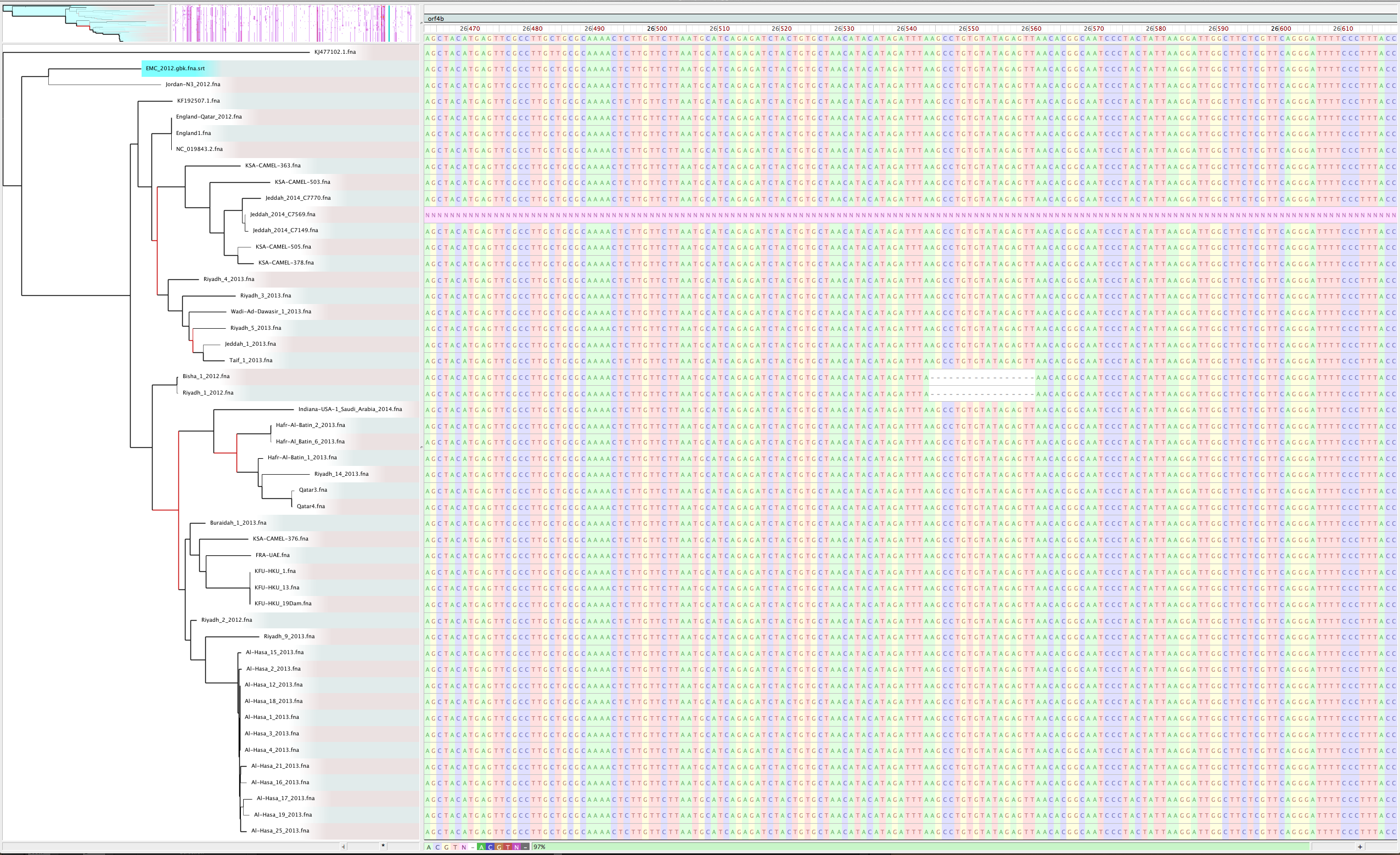
Zoom in with Gingr for nucleotide view of region
- On closer inspection, a large stretch of N’s in Jeddah isolate C7569 was the culprit
Inspect Output:
31 Streptococcus pneumoniae genomes
Download genomes:
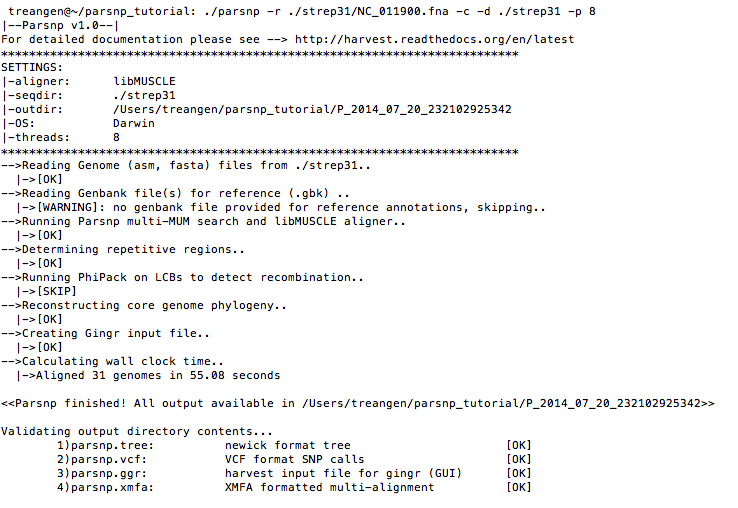
Run parsnp
parsnp -r ./strep31/NC_011900.fna -d ./strep31 -p <num threads>Command-line output:
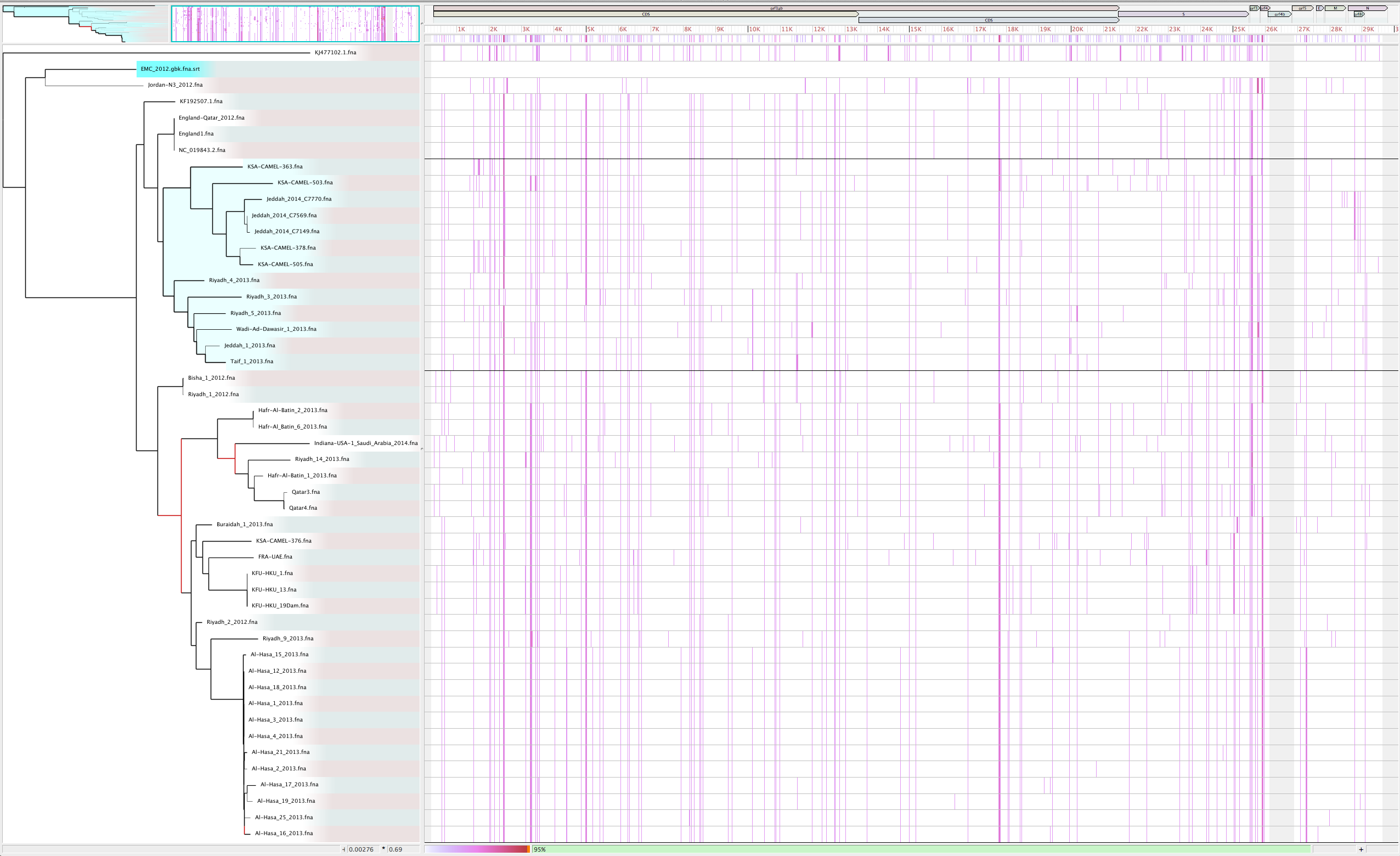
Force inclusion of all genomes (-c)
parsnp -r ./strep31/NC_011900.fna -d ./strep31 -p <num threads> -c
Command-line output: